Paper review 7
Biologically informed deep neural network for prostate cancer discovery, Nature [Paper]
Summary
- Biologically informed deep neural network (using Reactome pathway hierarchy)
- Better than baseline models, but suspicious of the way they integrate genomics profiles
- What if integrating GWAS?
Introduction
The advances in sparse model development and attribution methods have informed the development of deep learning models to solve biological problems using customized neural network architectures that are inspired by biological systems.
For example, visible neural networks were developed to model the effect of gene interaction on cell growth in yeast (DCell) and cancer cell line interactions
with therapies (DrugCell). A pathway-associated sparse deep neural
network (PASNet) used a flattened version of pathways to predict
patient prognosis in Glioblastoma multiforme.
Here they hypothesized that a biologically informed deep learning model built on advances in sparse deep learning architectures, encoding of biological information and incorporation of explainability algorithms would achieve superior predictive performance compared with established models and reveal novel patterns of treatment resistance in prostate cancer, with translational implications.
They developed P-NET—a biologically informed deep learning model—to stratify patients with prostate cancer by treatment-resistance state and evaluate molecular drivers of treatment resistance for therapeutic targeting through complete model interpretability.
From Genome to phenome.
Method
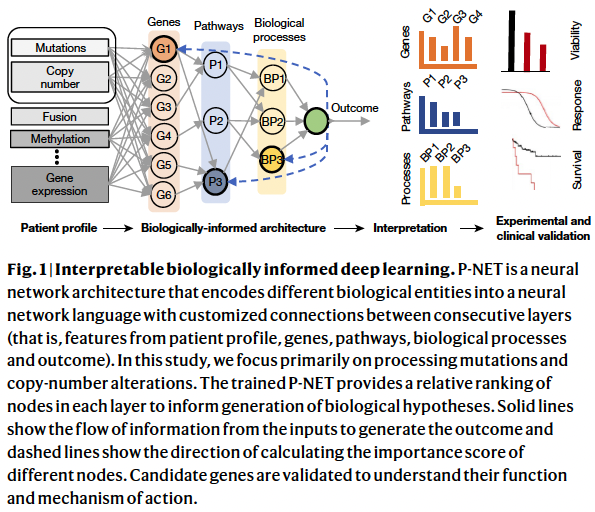
In P-NET, the molecular profile of the individual is fed into the model and distributed over a layer of nodes representing a set of genes using weighted links (Fig. 1, Extended Data Fig. 1). Later layers of the network encode a set of pathways with increasing levels of abstraction, whereby lower layers represent fine pathways and later layers represent more complex biological pathways and biological processes.
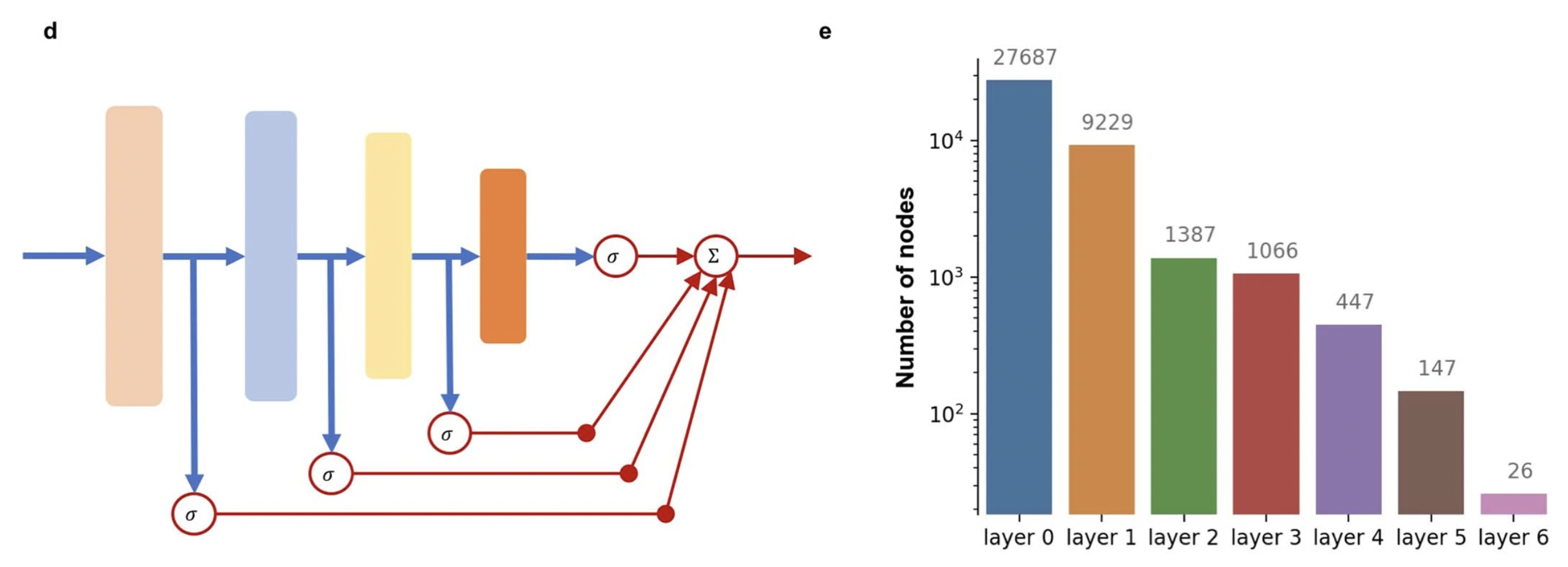
The architecture was built using the Reactome pathway datasets. The whole Reactome dataset was downloaded and processed to form a layered network of five layers of pathways, one layer of genes, and one layer for features.
layer 0: mutations(0/1)+copy number amplification(>1.0)+copy number deletions (27687)
layer 1: gene (9229)
layer 2: low-level pathway (1387)
layer 3: … (1066)
layer 4: … (447)
layer 5: … (147)
layer 6: high-level pathway(26) \
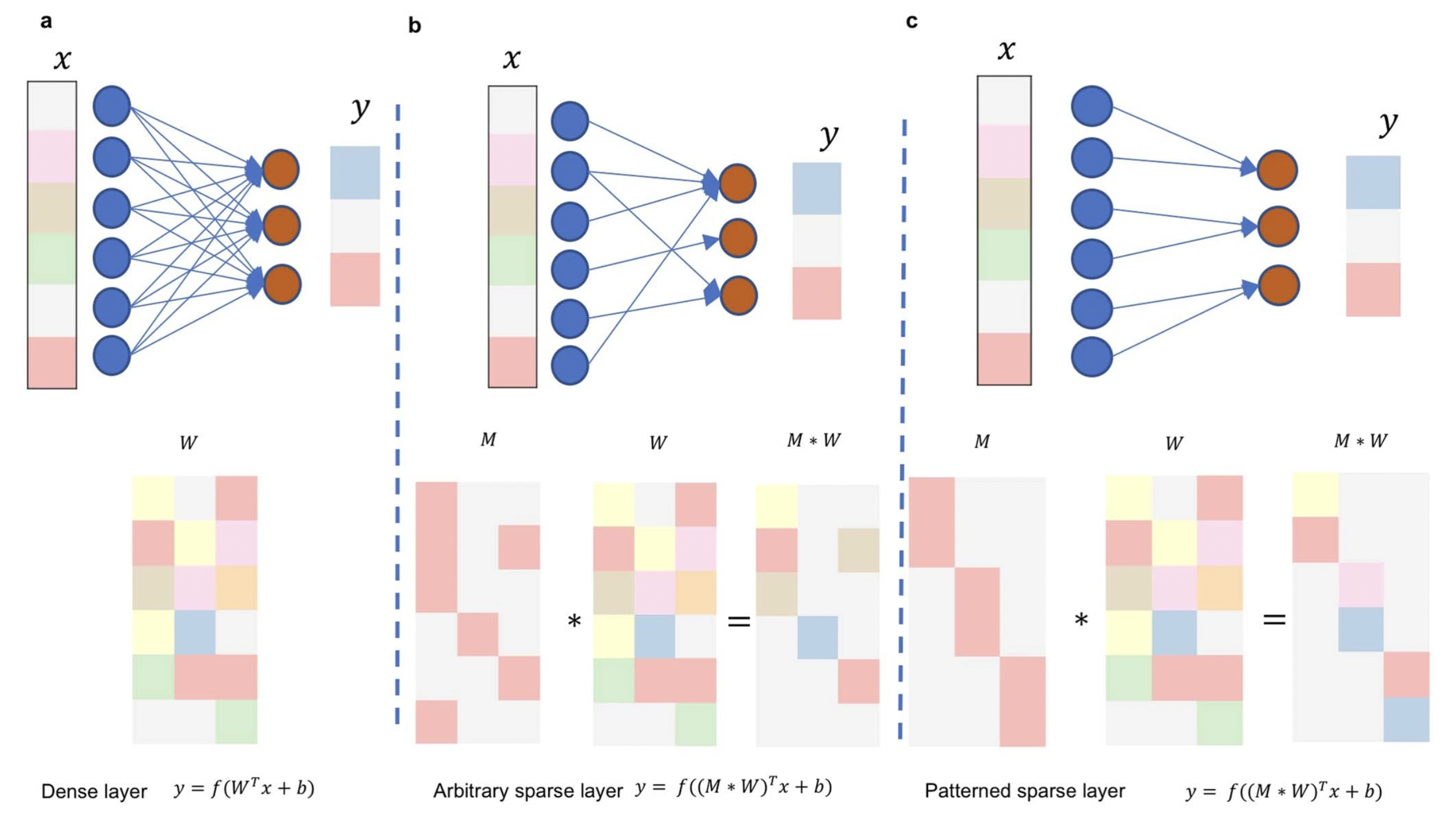
W_0: one-to-one connection
W_1: gene-pathway relationships from Reactome
W_2, W_3, W_4, W_5, W_6: from Reactome hierarchy
For Dense network, for example,
n(W_0) = (27687+1)9229 = 255m
n(W_1) = (9229+1)1387 = 12m
n(W_2) = (1387+1)*1066 = 1.4m
To allow each layer to be useful by itself, we added a predictive layer with sigmoid activation after each hidden layer. Since it is more challenging to fit the data using a smaller number of weights in the later layers, we used a higher loss weight for later layer outcomes during the optimization process. Since it is more challenging to fit the data using a smaller number of weights in the later layers, they used a higher loss weight for later layer outcomes during the optimization process.
Interpretation
To rank the features in all the layers, they chose to use the Deep-LIFT scheme as implemented in the DeepExplain library.
Training and Evaluation
The mutations were aggregated on the gene level with focus on nonsynonymous mutations to align with prior work on mutational significance in prostate cancer whole-exome datasets, excluding silent, intron, 3′ untranslated region (UTR), 5′ UTR, RNA and long intergenic non-coding RNA (lincRNA) mutations.
The copy number alterations for each gene were assigned on the basis of the called segment-level copy number emphasizing high gains and deep deletions and excluding single-copy amplification and deletions, as defined by GISTIC2.0 and generated from the source data type.
Transcriptomes (Fusion, Expression) To test model flexibility for RNA-based fusion inputs, as a secondary analysis we also developed P-NET models trained to predict cancer state incorporating fusions or different definitions of copy number states
(train 80% val 10%)*five fold, test 10%
To mitigate the bias issue in the main dataset, we trained two models on two balanced subsamples drawn from the main dataset. The prediction scores of the two models are averaged to produce the final predictions on the two external validation datasets.
Results
Internal set: 1013 samples (333 metastatic, 680 primary) (Armenia et al.) External validation (Fraser et al. + Robinson et al.)
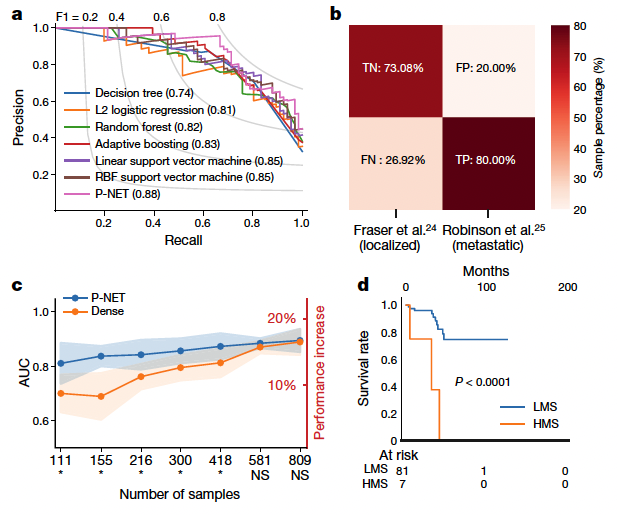
Incorporation of additional molecular features was feasible in P-NET (for example, fusions) but did not impact the performance of the model in this specific prediction task (Extended Data Figs. 3, 4).
P-NET achieves better performance (measured as the average AUC over five cross-validation splits) with smaller numbers of samples compared to a dense fully connected network with the same number of parameters.
Furthermore, a dense network that had the same number of neurons and layers as P-NET but
a much larger number of parameters (14 million) also achieved inferior performance.
External validation: 73% true-negative rate (TN) and 80% true-positive rate (TP),
High score group vs low score group
high score group is more likely to have biochemical recurrence.
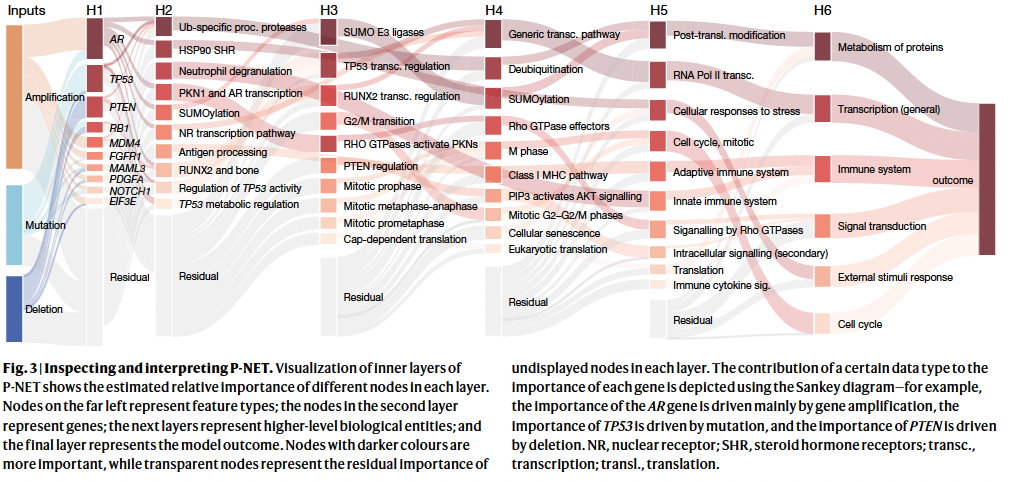
Interpretation - skip
Validation for interpretation - skip
Question
- mutations for each gene? # of mutations?