Paper review 3
Chemical-Reaction-Aware Molecule Representation Learning, arxiv (2021) [Paper]
Summary
- Impicitly learn Reaction template
- Training strategy: new contrastive learning loss (minimize pos pair of [R,P], and maximize neg pair of [R,P])
- One big limitation is NOT considering reaction conditions (temperature, pressure, catalysts, solvent, etc)
This is a super impressive paper! The idea was really elaborately designed and thoroughly followed basic organic chemistry (Highly generalized). Even, the experimental results are significantly improved.
Introduction
Molecule representation learning (MRL) tasks
- chemical reaction prediction
- molecule property prediction
- molecule generation
- drug discovery
- retrosynthesis planning
- chemical text mining
- chemical knowledge graph modeling
Previous works
1) natural language models: MolBERT, ChemBERTa, SMILES-Transformer, SMILES-BERT, Molecule-Transformer, SA-BiLSTM
Limitation: use SMILES, which simply based on “slender” strings.
2) GNN-based method
Jin et al., 2017; Gilmer et al.,2017; Ishida et al., 2021
Limitation: no generalization ability
Main idea
Proposes chemical reactions to assist learning molecule representations and improving their generalization ability.
A chemical reaction usually indicates a particular relation of equivalence between its reactants and products (e.g., in terms of conservation of mass and conservation of charge), and our idea is to preserve this equivalence in the molecule embedding space.
Two very nice properties
(1) Molecule embeddings are composable with respect to chemical reactions, which
make the embedding space well-organized (see Proposition 1);
(2) More importantly, it will show later that, when the molecule encoder is a GNN with summation as the readout function, our model can automatically and implicitly learn reaction templates that summarize a group of chemical reactions within the same category (see Proposition 2).
The ability of learning reaction templates is the key to improving the generalization ability of molecule representation, since the model can easily generalize its learned knowledge to a molecule that is unseen but belongs to the same category as or shares the similar structure with a known molecule.
NOT consider Reaction conditions (temperature, pressure, catalysts, solvent, etc) in this work.
MolR achieves 17.4% absolute Hit@1 gain in chemical reaction prediction, 2.3% absolute AUC gain on BBBP dataset in molecule property prediction, and 18.5% relative RMSE gain in graph-edit-distance prediction.
Method
Structural molecule encoder
Initial representation
atom $ a_i $ : concatenation four one-hot vectors [element type, charge, whether the atom is an aromatic ring, the count of attached hydrogen atoms] (each one-hot vector includes “unknown” entry)
bond $ b_i $ : bond type (e.g., single, double) not used. Since the bond type can usually be inferred by the features of its two associated atoms and does not consistently improve the model performance according to our experiments, it does not explicitly take bond type as input.
Preserving chemical reaction equivalence
A key idea is to preserve such equivalence in the molecule embedding space (R is a set of reactant, P is a set of product) $\sum_{r \in R}h_r = \sum_{p \in P}h_p$

reaction center of R -> P is defined as an induced subgraph of reactants R, in which each atom has at least one bond whose type differs from R to P (“no bond” is also seen as a bond type). In other words, a reaction center is a minimal set of graph edits needed to transform reactants to products.

the residual between reactant embedding and product embedding will fully and only depend on atoms that are less than hops away from the reaction center K.
general chemical reaction, reaction template, which abstracts a group of chemical reactions within the same category
For example,
(proved in Appendix C)
the residual between reactant embedding and product embedding will totally depend on the reaction center (colored in orange) as well as atoms whose distance from the reaction center is 1 or 2 (colored in light orange
Remarks
1) The model is theoretically able to learn a reaction template based on even only one reaction instance, which makes it particularly useful in few-shot learning scenario.
2) skip
3) K (# layers) can greatly impact the learned reaction templates. Sensitive to K
Training the model
Bad senario: all-zero embedddings for all molecule.
Common solutions to this problem are introducing negative sampling strategy or contrastive learning. Here we use a minibatch-based contrastive learning framework similar to CLIP since it is more time- and memory-efficient. But different from CLIP loss.
FYI, CLIP use average cross-entropy loss for dim=0 and dim=2 above.
Experiments
Chemical reaction prediction
Dataset, Zheng et al. (2019a), the dataset contains 478,612 chemical reactions as SMILES, and is split into training, validation, and test set of 408,673, 29,973, and 39,966 reactions, respectively, so we refer to this dataset as USPTO-479k.
Evaluation Protocol, task as ranking problem (all products in the test set as a candidate pool C, total 39,459 candidates). Rank by d(R, c).
MMR (mean reciprocal rank), MR (mean rank), Hit@1,3,5,10 as metrics. Repeated 3 times.
Baselines
MolBERT, Mol2vec (used the released pretrained model to ouput embeddings of reactants and cadidate products, and $ d(R,c)=-h_R^T h_c $)
Finetuning on USPTO-479k
- Mol2vec-FT1, MolBERT-FT1: train a diagonal matrix K $ d(R,c)=-h_R^T K h_c $\
- MolBERT-FT2: finetune by minimizing the contrastive loss function (eq. 5) on USPTO-497k
Hyperparameters setting, layers = [GCN, GAT, SAGE, TAG], K=2, output dimension=1024, margin=4, 20 epoch, batch size=4096, Adam optimizer, lr=10^-4
** TAG : Topological Adaptive Graph Convolutional Networks, a single layer aggregate from neighbors up to L hops away.
** Therefore, the actual number of layers for TAGCN is $ L^K $, which is $2^2 = 4 $ in the experiments.
Result
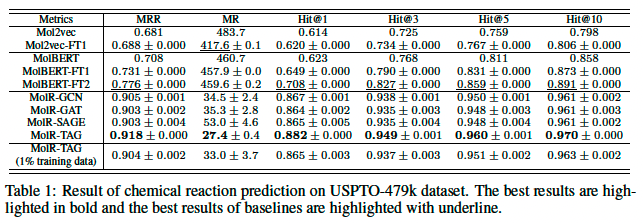
MolR shows thr best performance.
It performed well in few-show learning scenario.
Hyperparameter tuning by validation set
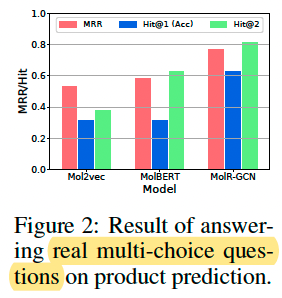
Real multi-choice questions, 16 questions on product prediction.
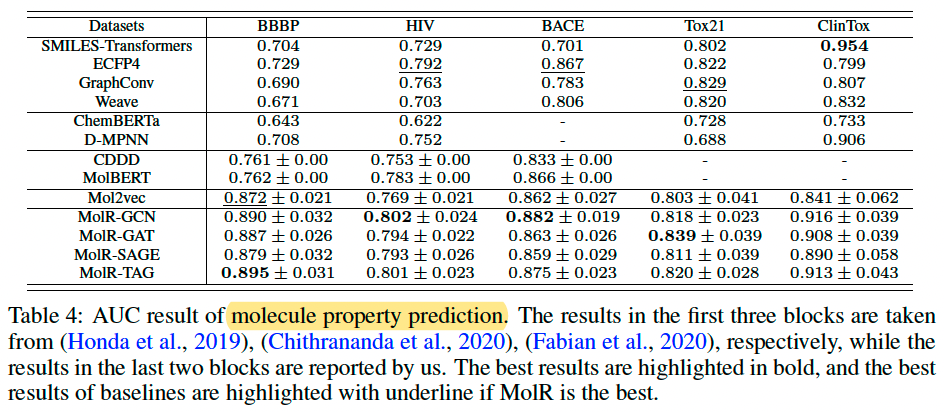
Molecule property prediction
Dataset, BBBP, HIV, BACE, Tox21, and ClinTox from Wu et al. (2018)
Each dataset contains thousands of molecule SMILES as well as binary labels indicating the property of interest (e.g., Tox21 measures the toxicity of compounds).
Baselines SMILES-Transformers, ECFP4, GraphConv, Weave, ChemBERTa, D-MPNN, CDDD, MolBERT, and Mol2vec
Experimental Setup, training, validation, and test set by 8:1:1. For MolR, used the model pretrained on USPTO-479k to process all datasets and output molecule embeddings, then feed embeddings and labels to a Logistic Regression model. Repeated 20 times.
Result
Graph-edit-distance prediction
Graph-edit-distance (GED) is a measure of similarity between two graphs, which is defined as the minimum number of graph edit operations to transform one graph to another. Predicts GED (structural similarity).
Experimental setup, randomly sample 10,000 molecule pairs from the first 1,000 molecules in QM9 dataset (Wu et al. (2018)), then calculate their ground-truth GEDs using NetworkX.
The dataset is split into training, validation, and test set by 8:1:1.
The embeddings of a pair of molecules are concatenated or subtracted as the features, and the ground-truth GEDs are set as targets, which are fed together to a Support Vector Regression model. Repeated 20 times,
Result
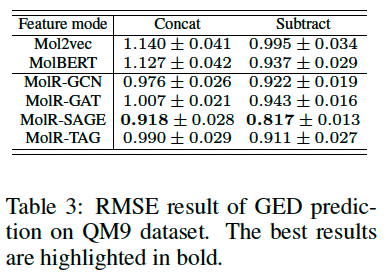
The result indicates that MolR can serve as a strong approximation algorithm to calculate GED. (ground-truth GEDs is [1, 14])
Embedding Visualization
Visualized the output embeddings of molecules in BBBP dataset by the pretrained MolR-GCN model, then visualize them using t-SNE.
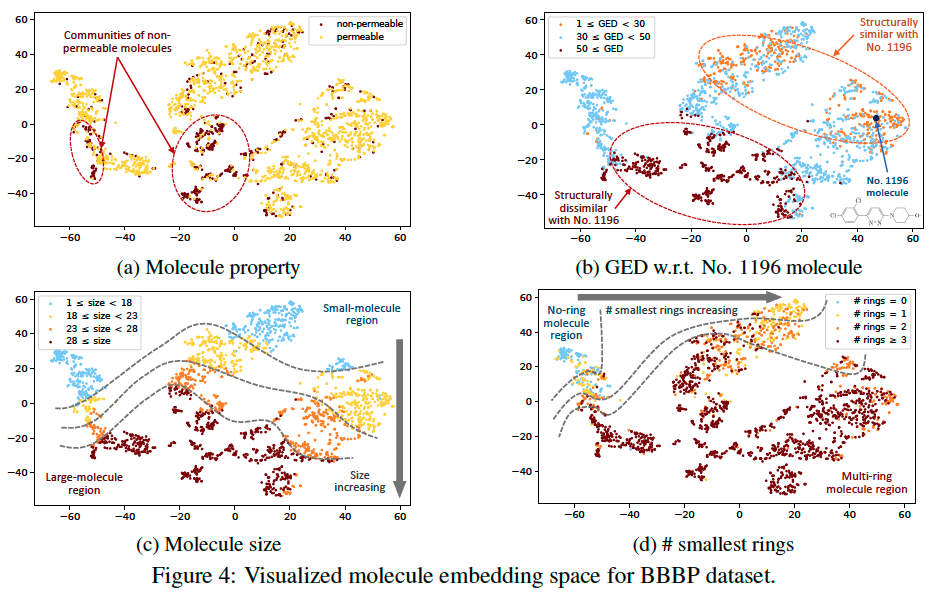
MolR encodes chemical reactions, for exmaples, alcohol oxidation and aldehyde oxidation.
 \
Visualize twi reactions, notice the direction and magnitude.
\
Visualize twi reactions, notice the direction and magnitude.
Note that the length of blue arrow is about twice as much as the corresponding red or orange arrow, which is exactly because (CH2OH)2/(CH2CHO)2 has two hydroxyl/aldehyde groups to be oxidized.
Related work
MolBERT uses BERT as the base model and designs three self-supervised tasks to learn molecule representation. However, SMILES is 1D linearization of molecular structure and highly depends on the traverse order of molecule graphs. This means that two atoms that are close in SMILES may actually be far from each other and thus not correlated.
Mol2vec treats molecule substructures (fingerprints) as words and molecules as sentences, then uses a Word2vec-like method to calculate molecule embeddings.
GNN-based methods (Jin et al., 2017; Gilmer et al., 2017; Ishida et al., 2021) usually require a large volume of training data due to their complicated architectures, which may limit their generalization ability when the data is sparse
Future work
- environmental condition
- explicitly output learned reaction templates
- how to differentiate stereoisomers
- additional information (e.g., textual description of molecules)
My Question
- how to convert SMILE to graph?
- how to find C?