Paper review 1
UNIFYING LIKELIHOOD-FREE INFERENCE WITH BLACK-BOX SEQUENCE DESIGN AND BEYOND [Paper]
Summary
- New de novo biological sequence design algorithm for a limited evaluation process
- Based on black-box optimization formulation
- Performed on TF binding sites, 5’ UTR, Antimicrobial peptides, and Fluorescence proteins.
[Caution!] I have no deep background on Likelihood-free inference (LFI) and black-box optimization problem at all. Just followed the paper’s introduction of them.
Introduction and Background
De novo biological sequence design is challenging for two reasons:
(1) the exploration space for sequences is combinatorially large
(2) sequence usefulness is evaluated via a complicated process which usually involves time-consuming and expensive wet-lab experiments
Likelihood-free inference (LFI)
LFI refers to a special kind of Bayesian inference setting where the likelihood function is not tractable but sampling (by simulation) from the likelihood is feasible.
While we do not have access to the exact likelihood, we can still simulate (sample) data from the model simulator. LFI only tries to obtain an approximation of the posterior for the given data.
Biological black-box sequence design
f(.) is the oracle score function. discover values of m for which f(m) is large.
In real-world situations, a query of this oracle f could represent a series of wet-lab experiments to measure specific chemical properties or specificity for a given binding site target. In general, these experiments are time- and cost-consuming. As a result, the total number of queries is limited.
Connecting LFI and sequence design
Think of $\varepsilon$ as a Boolean feature of these desirable configurations, and our goal is to characterize the posterior distribution $p(m|\varepsilon)$ from which we obtain the desired sequences.


Notice that in sequence design, the oracle could be either exact or noisy, thus we use the more general $s \sim f(m)$ formulation rather than $s = f(m)$.
Methods
Backward modeling
FB-VAE Gupta & Zou (2019)
It would then be natural to construct an analogical sequence design algorithm using top scored entities ${m_i}i$ to guide the update of a certain sequence distribution.

$q{\phi}(m)$ is variational autoencoder.
DbAs Brookes & Listgarten (2018)

fitting $q_{\phi}(m)$ through minimizing the KL divergence with the posterior $p(m|\varepsilon)$
Forward modeling
Iterative Scoring
Train a regressor $\hat{f_{\phi}}(m)$ in a supervised manner to fit the oracle scorer. Use $\tilde{q}(m)$ to denote the unknown posterior $p(m|\varepsilon)$ with knowledge of $\hat{f_{\phi}}(m)$ and prior $p(m)$.

If choose Example A to serve as the definition of event $\varepsilon$, then the samples of $\tilde{q}(m)$ can be obtained in this way: (1) sample m from prior $p(m)$ and (2) accept this sample if $\hat{f_{\phi}}(m)$ is larger than threshold s, or otherwise reject it.
Alternatively, if we choose Example B, we have $p(m) exp(\hat{f_{\phi}}(m) /\tau)$.
Modeling a probability ratio
Iterative Ratio

Details are skipped
Composite probabilistic methods
Iterative Posterior Scoring and Iterative Posterior Ratio
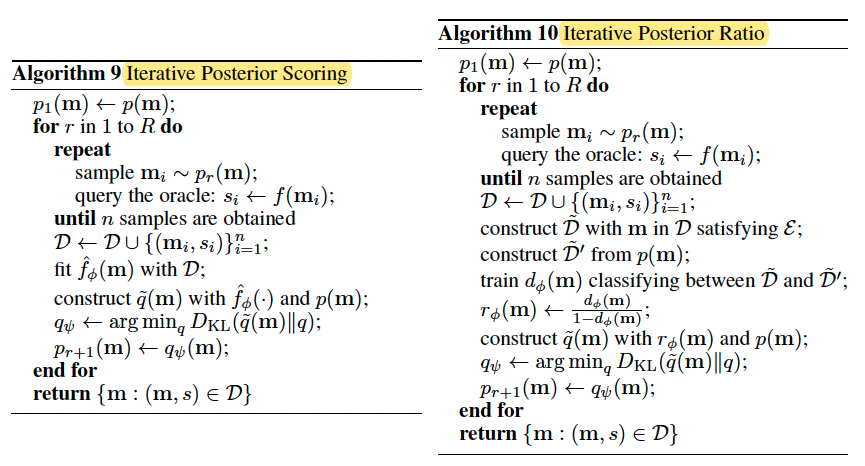
Details are skipped
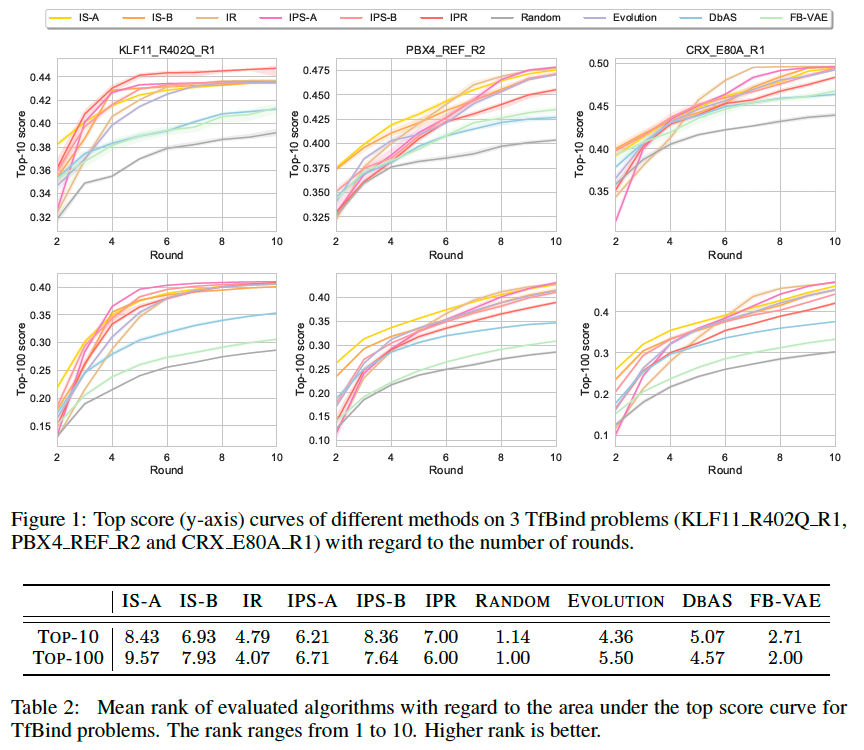
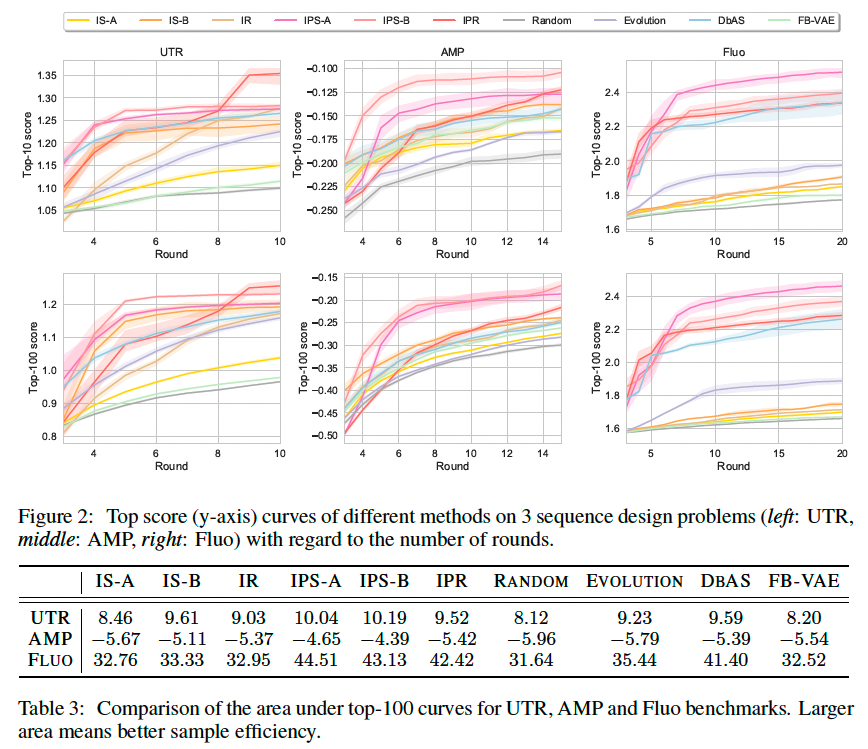
Experiments and Results
Datasets
Transcription factor binding sites (TfBind)
In Barrera et al. (2016), the authors measure the binding properties between a battery of transcription factors and all possible length-8 DNA sequences through biological experiments.
5’ untranslated regions (UTR)
In Sample et al. (2019), the authors create a library of gene sequences with ribosome loading level as labels. They further train a convolutional neural network with this library to predict the relationship between a 5’UTR sequence and the corresponding gene expression level. Use this neural network as an oracle for this benchmark. The length of the gene sequences is fixed to 50.
Antimicrobial peptides (AMP)
Use the AMP dataset from (Witten & Witten, 2019) which contains 6,760 AMP sequences. A multilayer perceptron classifier is trained to predict if a protein sequence can prohibit the growth of a particular pathogen in that AMP dataset. This classifier operates on the features extracted by ProtAlbert (Elnaggar et al., 2020) model. The length of sequences ranges from 12 to 60.
Fluorescence proteins (Fluo)
Ground-truth measurement relies on a regressor. Use a pretrained model taken from Rao et al. (2019) to act as the task oracle, which is a regressor trained to predict log-fluorescence intensity value over approximately 52,000 protein sequences of length 238 (Sarkisyan et al., 2016).
Model details
The encoder of the VAE first linearly transform one-hot input into a hidden feature which is 64 dimension, and then separately linearly transform to a 64-dimension mean output and 64-dimension variance output. The decoder contains a 64 64 linear layer and a linear layer that maps the hidden feature to categorical output. All other methods utilize bi-directional long short-term memory model (BiLSTM) with a linear embedding layer. Both the embedding dimension and the hidden size of LSTM is set to 32.
Result
Evaluate these sequence design algorithms by the average score of the top-10 and top-100 sequences in the resulting dataset D at each round.